| TUD Organische Chemie | Immel | Tutorials | Orbitals | Molecular | 1,3-Butadiene | View or Print (this frame only) |
The molecular orbitals (MOs) of molecules can be constructed by linear combination of atomic orbitals (LCAO).
Though the exact Schrödinger equation is unsolvable for many electron systems such
as molecules, the solution can be numerically approximated by ab initio or density functional (DFT) theory.
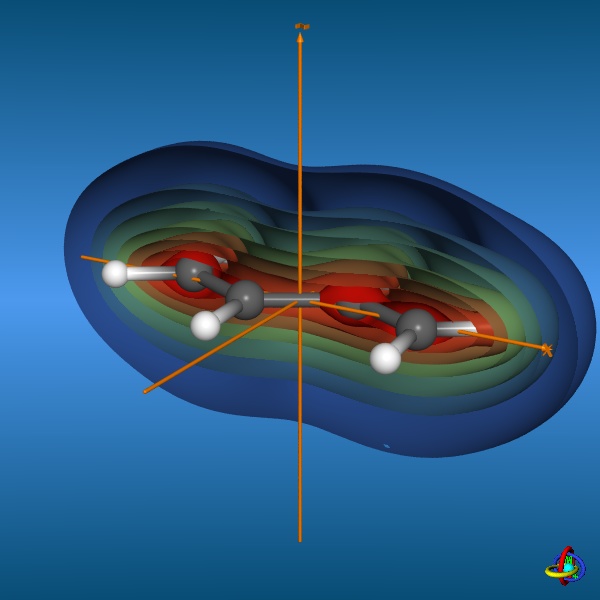
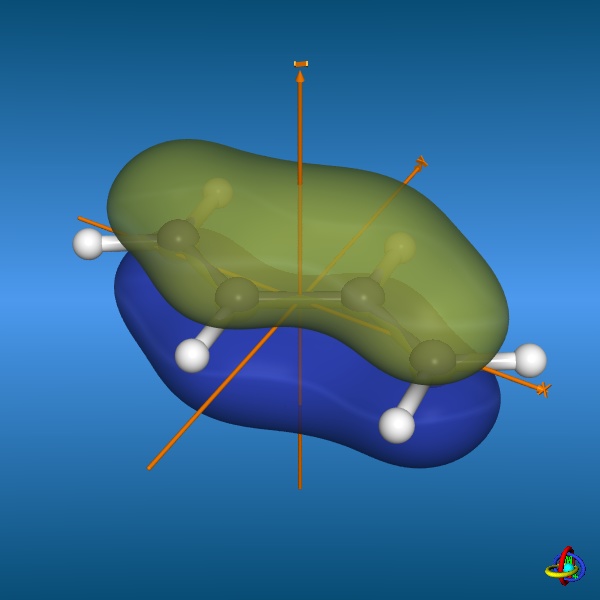
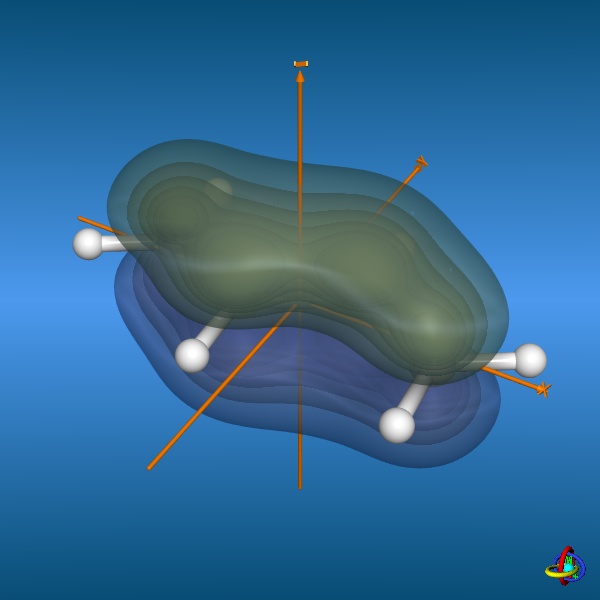
This page gives an overview on the molecular orbitals of butadiene calculated by DFT methods using a B3LYP/6-311++G** basis set. All MO representations are 90% or 90-25% iso-contour probability surfaces of the electron density (ψ2), i.e. they resemble the spatial volume around the nuclei of the molecule in which the electrons are found with the corresponding certainty. The different colors (yellow and blue) represent regions with opposite sign of the wave function ψ; nodal planes (not necessarily real "planar" planes) were ψ passes through zero and changes sign are indicated in orange.
Click on the small images below with blue background below to obtain an enlarged view - the images with black background provide links to the corresponding 3D-models (VRML-type models); these links will open in a new window. |
|
||||||||||||||||||||||||||||||||||||||||||||
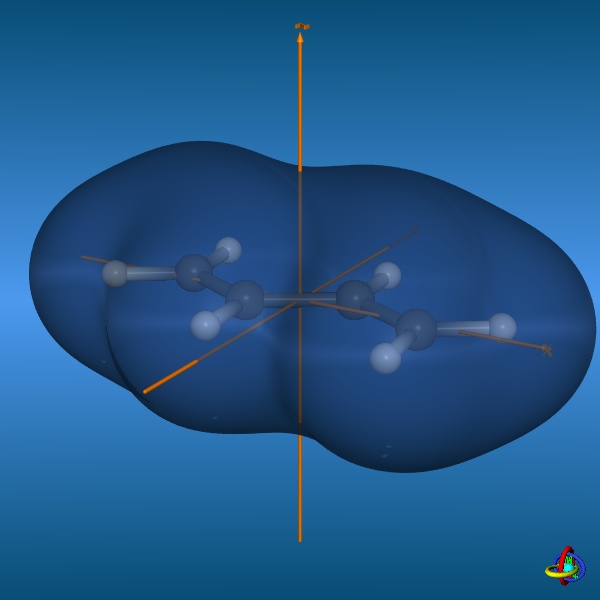
The total electron density (clipped 99, 95, 90, 80, 70, 60, and 50% iso-density contours depicted on the right) render the molecule with its characteristic shape
(note the different iso-contour values of the MO orbitals and the total electron density contours).
|
|
||||||||||||||||||||||||||||||||||||||||||||
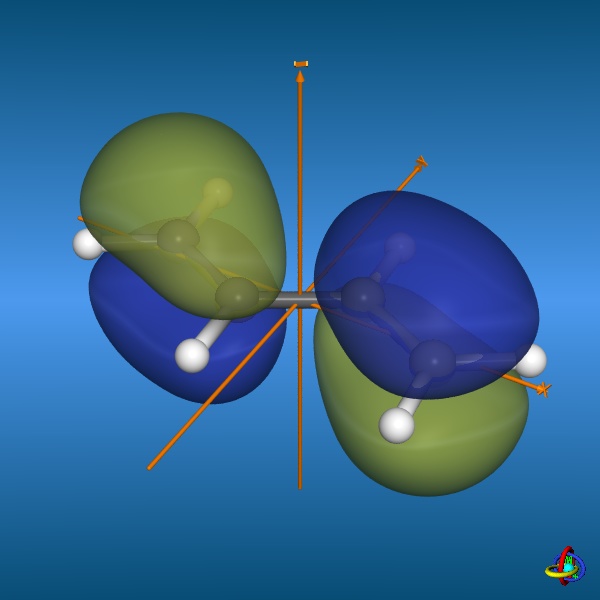
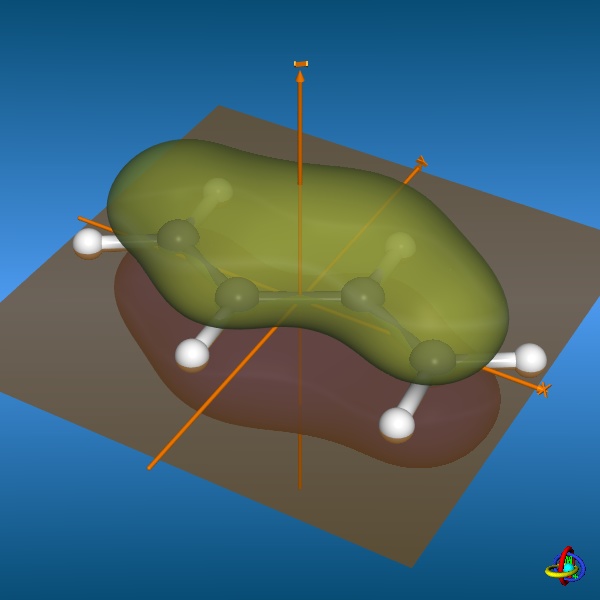
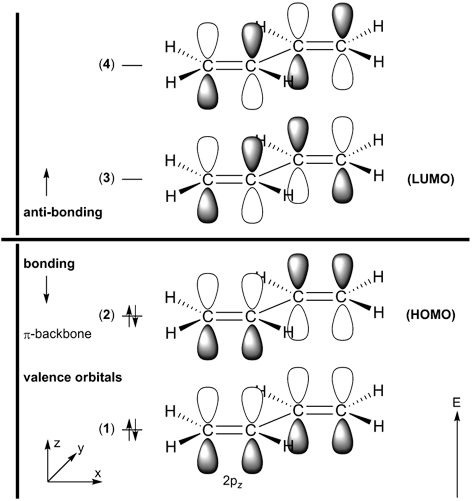
Below, three (out of four) π-molecular orbitals of butadiene are displayed. Similarly to the π-orbitals of ethene, these are made of the carbon
2pz orbitals with changing signs of the wave function ψ. The fully conjugated,
lowest-energy π-orbital (orbital no. 1) accounts for the partial double bond character of the central carbon-carbon bond, whilst the
highest occupied molecular orbital (HOMO, orbital no. 2) corresponds to two isolated double bonds.
Furthermore, the lowest unoccupied molecular orbital (LUMO, orbital no. 3) features an additional nodal plane.
The thumbnail images on the left provide access to enlarged graphics as well as 3D-models (VRML) of the orbitals, respectively. Further informations on AOs are available from the gallery of hydrogenic orbitals and hybrid orbitals. |
|||||||||||||||||||||||||||||||||||||||||||||
Notes: a) Orbital number (π-valence orbitals only, see scheme on the right, numbers of unoccupied orbitals are in italics); b) Label (symmetry descriptor in parenthesis); c) Nodal planes (ψ = 0.0); d) 90% Probability contours of MO electron density (ψ^2); e) 90, 80, 70, 60, 50, 40, and 25% Probability contours; f) 3D-Models require a VRML plugin to be installed (large files with sizes between 1000-3700 KBytes); g) Orbital not shown. |
|
||||||||||||||||||||||||||||||||||||||||||||
For a more detailed description of atomic orbitals see the corresponding gallery of orbitals.
All graphics and iso-contour surfaces shown on this page were created using the MolArch+ program
and POVRAY Persistence of Vision Raytracer. Electron densities were calculated
on three dimensional grids for the corresponding molecules using the JAGUAR program.
|
|||||||||||||||||||||||||||||||||||||||||||||